


PHARMAKOLOGIE
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Ranitidin: Rückruf von ranitidinhaltigen Arzneimitteln
Bonn (17. September 2019) — Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) informiert darüber, dass in der Europäischen Union ein Rückruf von Arzneimitteln erfolgt, die den von dem Wirkstoffhersteller Saraca Laboratories Limited hergestellten Wirkstoff Ranitidin enthalten. Es liegen Indizien vor, dass auch der Wirkstoff weiterer Wirkstoffhersteller von der Verunreinigung betroffen sein könnte. Ranitidinhaltige Arzneimittel
WEITERLESEN »
Wirkstoffe Acitretin, Adapalen, Alitretinoin, Bexaroten, lsotretinoin, Tazaroten, Tretinoin: Rote-Hand-Brief zu Retinoiden: Teratogenität und neuropsychiatrische Erkrankungen
Rote-Hand-Brief zu Retinoiden: Teratogenität und neuropsychiatrische Erkrankungen Bonn (9. September 2019) — Die Zulassungsinhaber von retinoidhaltigen Arzneimitteln informieren über Aktualisierungen zu Teratogenität und neuropsychiatrischen Erkrankungen. Retinoide sind stark teratogen und dürfen während der Schwangerschaft nicht angewendet werden. Bei der Anwendung oraler Retinoide bei Frauen im gebärfähigen Alter müssen alle Bedingungen des Schwangerschaftsverhütungsprogramms erfüllt werden.
WEITERLESEN »
IQWiG: Ezetimib senkt das Risiko für Herzinfarkte und Schlaganfälle
Köln (26. Juli 2019) — Additiv zu einer bestehenden Statintherapie gegeben, wirkt Ezetimib bei Patienten mit KHK und akutem Koronarsyndrom vorbeugend. Bei Patientinnen und Patienten mit koronarer Herzkrankheit (KHK) oder akutem Koronarsyndrom (ACS) in der Vorgeschichte ist es von höherem Nutzen, mit einem Statin in Kombination mit Ezetimib behandelt zu werden, als mit einem
WEITERLESEN »
Kardio-renale Patienten: Veltassa® – Effektives und interdisziplinäres Management der Hyperkaliämie im Praxisalltag.
Veltassa® – Effektives und interdisziplinäres Management der Hyperkaliämie im Praxisalltag München (19. Juli 2019) — Kardio-renale Patienten weisen im Behandlungsalltag häufig erhöhte Serumkaliumwerte (Hyperkaliämie) auf, nicht zuletzt, wenn ein Einsatz von Inhibitoren des Renin-Angiotensin-Aldosteron-Systems (RAASi) gemäß der Leitlinien erfolgt.1 Dies führt oft zum Absetzen oder zur Dosisreduktion der RAASi, was mit einem erhöhten Risiko
WEITERLESEN »
Rote-Hand-Brief zu elmiron® (Pentosanpolysulfat-Natrium): Risiko von pigmentärer Makulopathie
Bonn (18. Juli 2019) — Die Firma bene-Arzneimittel GmbH informiert in Abstimmung mit der Europäischen Arzneimittel-Agentur (EMA) und dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über seltene Fälle von pigmentärer Makulopathie unter der Anwendung von Pentosanpolysulfat-Natrium, die insbesondere nach Langzeitanwendung auftraten. Während der Behandlung sollten die Patienten zur Früherkennung einer pigmentären Makulopathie regelmäßig augenärztlich
WEITERLESEN »
Cyproteron: Überprüfung des Meningeomrisikos
Bonn (12. Juli 2019) — Die EMA hat mit der Überprüfung cyproteronhaltiger Arzneimittel begonnen, die zur Behandlung einer Reihe von Erkrankungen verwendet werden, darunter übermäßiges Haarwachstum, Prostatakrebs und Akne. Es wird auch in der Hormonersatztherapie angewendet. Im Rahmen des Risikobewertungsverfahrens soll das Risiko der Entwicklung eines Meningeoms untersucht werden, einem seltenen, meist nicht-bösartigen Tumor
WEITERLESEN »
Methotrexat: Dosierungsfehler
PRAC empfiehlt neue Maßnahmen zur Vermeidung von Dosierungsfehlern bei Methotrexat Bonn (12. Juli 2019) — Der Sicherheitsausschuss der EMA (PRAC) empfiehlt neue Maßnahmen zur Vermeidung von Dosierungsfehlern. Dosierungsfehler haben dazu geführt, dass einige Patienten fälschlicherweise jeden Tag, statt einmal pro Woche, methotrexathaltige Arzneimittel eingenommen haben. Zu den neuen Maßnahmen gehören die Einschränkung, dass
WEITERLESEN »
ViiV Healthcare erhält die europäische Zulassung für Dovato (Dolutegravir/Lamivudin), ein neues, einmal tägliches 2-Drug Regimen für die HIV-1 Therapie
Die Zulassung basiert auf den Daten der GEMINI-Studien, in denen Dolutegravir + Lamivudin im Vergleich mit einem Dolutegravir-basierten Dreifachregime zu Woche 48 virologisch nicht unterlegen war. Es kam nicht zur Entwicklung von Resistenzen. München (4. Juli 2019) – ViiV Healthcare hat mitgeteilt, dass die Europäische Kommission die Zulassung für Dovato (Dolutegravir/Lamivudin) zur Behandlung
WEITERLESEN »
IncobotulinumtoxinA (Xeomin®) schließt therapeutische Lücke bei unkontrolliertem Speichelfluss
Frankfurt am Main (2. Juli 2019) – Mit IncobotulinumtoxinA (Xeomin®) ist erstmals ein Botulinum Neurotoxin A-Präparat zur symptomatischen Behandlung der chronischen Sialorrhoe bei Erwachsenen infolge neurologischer Erkrankungen zugelassen.1 Nach Injektion in die Speicheldrüsen hemmt es reversibel und dosisabhängig die cholinerge neuroglanduläre Signalübertragung und damit die Aktivität der Speicheldrüsen, erklärte Professor Dr. med. Wolfgang Jost,
WEITERLESEN »
Omega-3-Fettsäuren: EMA bewertet die Anwendung nach Herzinfarkt
Bonn (18. Juni 2019) — Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) setzt mit Bescheid vom 14. Juni 2019 den entsprechenden Durchführungsbeschluss der Europäischen Kommission (2019) 4336 vom 06.06.2019 um. Damit wird das europäische Risikobewertungsverfahren nach Artikel 31 der Richtlinie 2001/83/EG zu „Omega-3-Säurenethylester“ (EMEA/H/A -31/1464) abgeschlossen. Das Gutachten des Ausschusses für Humanarzneimittel (CHMP) der
WEITERLESEN »
Leuprorelinhaltige Arzneimittel: Medikationsfehler bei Depotzubereitungen
Bonn (17. Juni 2019) — Die Europäische Arzneimittel-Agentur (EMA) überprüft leuprorelinhaltige Arzneimittel aufgrund von Berichten zu Medikationsfehlern, die auf Zubereitungs- und Applikationsfehler zurückzuführen waren. Dadurch wurde bei einigen Patienten eine zu geringe Menge des Arzneimittels appliziert, was den Behandlungserfolg beeinträchtigte. Diese Überprüfung umfasst Depotformulierungen, die als Injektion unter die Haut oder in den Muskel
WEITERLESEN »
Forschung, Wissenschaft & Therapie: Chiesi punktet mit 3-fachem Engagement als starker Partner in der Transplantationsmedizin
Hamburg (27. Mai 2019) — Die immunsuppressive Therapie nach Organtransplantation wurde in den letzten Jahren immer weiterentwickelt. Dennoch müssen noch viele Fragen geklärt und medical needs, die sich in der täglichen Praxis ergeben, erforscht werden. Chiesi unterstützt daher mit TACKLE, einem Programm zur Forschungsförderung, vor allem auch anwenderbetonte Projekte in Deutschland. Zudem bietet Chiesi
WEITERLESEN »
Wirkstoffe Apixaban, Dabigatranetexilat, Edoxaban, Rivaroxaban: Rote-Hand-Brief zu Eliquis®, Pradaxa®, Lixiana®/Roteas® und Xarelto®: Die Anwendung bei Patienten mit Antiphospholipid-Syndrom wird nicht empfohlen
Rote-Hand-Brief zu Eliquis®, Pradaxa®, Lixiana®/Roteas® und Xarelto®: Die Anwendung bei Patienten mit Antiphospholipid-Syndrom wird nicht empfohlen Bonn (23. Mai 2019) – Die Firmen Bayer AG, Boehringer Ingelheim International GmbH, Bristol-Myers Squibb/Pfizer EEIG und Daiichi Sankyo Europe informieren in Abstimmung mit der Europäischen Arzneimittel-Agentur (EMA) und dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über die
WEITERLESEN »
BfArM: Xeljanz® (Tofacitinib): Einschränkungen bei der Anwendung wegen des Risikos von Blutgerinnseln in der Lunge
Xeljanz® (Tofacitinib): Einschränkungen bei der Anwendung wegen des Risikos von Blutgerinnseln in der Lunge Bonn (17. Mai 2019) – Der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) bei der Europäischen Arzneimittel-Agentur (EMA) hat empfohlen, dass Ärzte die zweimal täglich zu verabreichende Dosis von 10 mg Xeljanz® (Tofacitinib) nicht bei Patienten verschreiben dürfen, die
WEITERLESEN »
Rote-Hand-Brief zu biotinhaltigen Arzneimitteln: Risiko falscher Ergebnisse von Laboruntersuchungen durch Biotininterferenzen
Bonn (15. Mai 2019) – Die Firmen, die biotinhaltige Arzneimittel sowie biotinhaltige Nahrungsergänzungsmittel oder diätetische Lebensmittel für besondere medizinische Zwecke vermarkten, informieren in Absprache mit dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über mögliche Interferenzen von Biotin mit klinischen Laboruntersuchungen (insbesondere Bestimmung von Biomarkern wie z.B. von Hormonen, Herz-, Tumor- oder Infektionsmarkern) und dem
WEITERLESEN »
Rote-Hand-Brief zu Modafinil: Mögliches Risiko schwerer angeborener Fehlbildungen
Bonn (9. Mai 2019) – Die Zulassungsinhaber von modafinilhaltigen Arzneimitteln informieren in Abstimmung mit dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über neue Erkenntnisse im Zusammenhang mit der Anwendung von Modafinil während der Schwangerschaft. Es besteht der Verdacht, dass die Anwendung von Modafinil während der Schwangerschaft zu schweren angeborenen Fehlbildungen führen kann. Modafinil sollte
WEITERLESEN »
Rote-Hand-Brief zu Domperidon: Erinnerung an Maßnahmen zur Minimierung kardialer Risiken
Bonn (29. April 2019) – Die Zulassungsinhaber domperidonhaltiger Arzneimitteln möchten Sie in Abstimmung mit dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) nochmals an die Sicherheitsmaßnahmen aus dem Jahr 2014 erinnern. Eine jetzt beendete Studie zur Überprüfung der Einhaltung dieser risikominimierenden Maßnahmen hat nun ergeben, dass Ärzte zum Teil nicht hinreichend mit den Empfehlungen zur
WEITERLESEN »
BfArM: Xeljanz® (Tofacitinib): Erhöhtes Risiko von Blutgerinnseln in der Lunge und Tod durch höhere Dosis bei rheumatoider Arthritis
Xeljanz® (Tofacitinib): Erhöhtes Risiko von Blutgerinnseln in der Lunge und Tod durch höhere Dosis bei rheumatoider Arthritis Berlin (20. März 2019) – Die Europäische Arzneimittelagentur (EMA) rät Ärzten und Patienten, die empfohlene Dosis von Xeljanz® (Wirkstoff: Tofacitinib) bei der Behandlung von rheumatoider Arthritis nicht zu überschreiten. Der Hinweis basiert auf frühen Ergebnissen einer laufenden
WEITERLESEN »
Fenspiridhaltige Arzneimittel: Potenzielles Risiko von Herzrhythmusstörungen
Bonn (15. Februar 2019) – Der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) hat eine EU-weite Aussetzung von fenspiridhaltigen Arzneimitteln empfohlen, die bei Kindern und Erwachsenen zur Linderung von Husten infolge von Lungenerkrankungen eingesetzt werden. Das Ruhen der Zulassung ist eine Vorsichtsmaßnahme zum Schutz der Patienten, während der PRAC das Risiko einer QT-Verlängerung
WEITERLESEN »

Wirkstoff Apixaban, Dabigatranetexilat, Rivaroxaban: Die EMA startet die Bewertung einer Studie zum Blutungsrisiko direkter oraler Antikoagulantien
London, UK (8. Februar 2019) – Die Europäische Arzneimittelagentur (EMA) prüft die Ergebnisse einer Studie mit den direkten oralen Gerinnungshemmern (direkte orale Antikoagulantien – DOAC) Eliquis® (Apixaban), Pradaxa® (Dabigatranetexilat) und Xarelto® (Rivaroxaban). Diese von der EMA in Auftrag gegebene Beobachtungsstudie untersuchte die Häufigkeit schwerer Blutungen dieser Arzneimittel im Vergleich zu anderen oralen Antikoagulantien (Vitamin
WEITERLESEN »
Rote-Hand-Brief zu carbimazol- oder thiamazolhaltigen Arzneimitteln: Risiko einer akuten Pankreatitis und Verstärkung der Empfehlung zur Kontrazeption
Berlin (6. Februar 2019) – Die Zulassungsinhaber carbimazol- und thiamazolhaltiger Arzneimittel informieren nach Abschluss eines europäischen Signalverfahrens in Abstimmung mit dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über das Risiko einer akuten Pankreatitis und die dringende Empfehlung, dass Frauen im gebärfähigen Alter während der Behandlung mit Carbimazol oder Thiamazol wirksame Methoden der Kontrazeption anwenden.
WEITERLESEN »
Aspirin, Ibuprofen- und Ibuprofen-Lysin-Tabletten im Vergleich: Neue Studienergebnisse bestätigen bessere Zerfallsgeschwindigkeit der Aspirin® Tablette erstmals in vivo
Aspirin zerfällt viermal schneller als Ibuprofen- und Ibuprofen-Lysin-Tabletten Aspirin erreicht seine maximale Plasmakonzentration dreimal schneller als Ibuprofen und zweimal schneller als Ibuprofen-Lysin Aspirin hat eine kurze Kontaktzeit mit der Magenschleimhaut und verlässt den Magen rasch Leverkusen (23. Januar 2019) – Aspirin ist weltweit eines der am besten erforschten Arzneimittel überhaupt.(1-3),[1], Die bereits vorhandenen,
WEITERLESEN »
Rote-Hand-Brief zu SGLT2-Inhibitoren („Sodium-Glucose-Co-Transporter 2 Inhibitors“): Risiko einer Fournier Gangrän (nekrotisierende Fasziitis des Perineums) bei der Anwendung
Bonn (21. Januar 2019) – Die Zulassungsinhaber von SGLT2-Inhibitoren informieren in Abstimmung mit der Europäischen Arzneimittel-Agentur (EMA) und dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) darüber, dass nach Markteinführung Fälle von Fournier Gangränen (nekrotisierende Fasziitis des Perineums) mit der Anwendung von SGLT2-Inhibitoren in Verbindung gebracht wurden. Die Fournier Gangrän ist eine seltene, aber schwere
WEITERLESEN »
Risdiplam erhält PRIME-Status der EMA zur Behandlung von Spinaler Muskelatrophie
Grenzach/Wyhlen (18. Januar 2019) – Die Europäische Arzneimittel-Agentur (EMA) hat den PRIority-MEdicines (PRIME)-Status für das orale Prüfmedikament Risdiplam (RG7916) von Roche zur Behandlung von Patienten mit Spinaler Muskelatrophie (SMA) erteilt. Der PRIME-Status der EMA wird vielversprechenden Arzneimitteln erteilt, um deren Entwicklungspläne und die Generierung von Daten zu fördern.1 Der Spleißmodifikator Risdiplam wird derzeit in
WEITERLESEN »
Rote-Hand-Brief zu Kybella® 10 mg/ml Injektionslösung (Deoxycholsäure): Risiko einer Nekrose an der Injektionsstelle
Kybella® 10 mg/ml Injektionslösung (Deoxycholsäure): Risiko einer Nekrose an der Injektionsstelle Bonn (17. Januar 2019) – Die Firma Allergan Pharmaceuticals International Limited informiert in Abstimmung mit der Europäischen Arzneimittel-Agentur (EMA) und dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) über Nekrosen an der Injektionsstelle, einschließlich Arteriennekrosen, im submentalen Behandlungsbereich oder in der unmittelbaren Umgebung bei
WEITERLESEN »
Envarsus® – neue Real-World-Daten mit dem retardierten Tacrolimus mit MeltDose®-Technologie zur Wirksamkeit und Sicherheit bei Leber- und Nierentransplantation
Hamburg (11. Dezember 2018) – Die immunsuppressive Therapie nach allogener Organtransplantation mit Tacrolimus ist klinischer Standard. Das retardierte TacrolimusPräparat Envarsus® weist ein verändertes pharmakokinetisches Profil mit verbesserter Bioverfügbarkeit auf, was zu stabilen, flacheren Wirkstoffspiegeln im Blut führt.1 Erfahrungen im Praxisalltag mit diesem Tacrolimus-Präparat nach Leber- und Nierentransplantation unterstreichen die Ergebnisse klinischer Studien zu Wirksamkeit
WEITERLESEN »
Rote-Hand-Brief zu dienogest- und ethinylestradiolhaltigen Kontrazeptiva: Risiko venöser Thromboembolien
Bonn (11. Dezember 2018) – Die Firma Jenapharm GmbH & Co. KG informiert über das Risiko venöser Thromboembolien (VTE) bei Frauen, die dienogest- und ethinylestradiolhaltige Kontrazeptiva anwenden, im Vergleich zu Anwenderinnen levonorgestrel- und ethinylestradiolhaltigen Kombinationen, die mit einem niedrigeren Risiko verbunden sind. Die Höhe des Risikos venöser Thromboembolien unter der Einnahme einer Kombination aus
WEITERLESEN »
BfArM: Fluorchinolone: Schwere und langanhaltende Nebenwirkungen im Bereich Muskeln, Gelenke und Nervensystem
Fluorchinolone: Schwere und langanhaltende Nebenwirkungen im Bereich Muskeln, Gelenke und Nervensystem Gutachten des CHMP Einschränkungen in der Anwendung aufgrund von möglicherweise dauerhaften und die Lebensqualität beeinträchtigenden Nebenwirkungen Zugelassene Wirkstoffe in Deutschland: Ciprofloxacin, Levofloxacin, Moxifloxacin, Norfloxacin, Ofloxacin Bonn (16. November 2018) – Die europäische Arzneimittelagentur EMA hat die schwerwiegenden, die Lebensqualität beeinträchtigenden und potentiell
WEITERLESEN »
Novartis bringt mit Aimovig® (Erenumab) die erste spezifisch entwickelte Migräneprophylaxe in Deutschland auf den Markt
Aimovig® (Erenumab) steht in Deutschland ab 1. November 2018 zur Prophylaxe der Migräne bei Erwachsenen mit 4 oder mehr Migränetagen pro Monat zur Verfügung.1 Einfache Anwendung mit dem SureClick® Fertigpen. Erenumab zeigte in klinischen Studien mit über 3.000 Patienten eine signifikante Wirksamkeit, eine Verträglichkeit auf Placebo-Niveau und einen schnellen Wirkeintritt, auch bei schwierig zu
WEITERLESEN »

Pelgraz® – erstes Pegfilgrastim Biosimilar in der EU sorgt für deutliche Einsparungen und vergrößert damit den Zugang zu diesem lebenswichtigen Medikament
Erstes pegyliertes G-CSF Biosimilar in der EU in der Neutropeniebehandlung zugelassen und bereits in Deutschland seit dem 25. September 2018 verfügbar Accord Healthcare bringt Pelgraz® (Pegfilgrastim) EU-weit auf den Markt Einzig verfügbares Pegfilgrastim-Biosimilar mit Wirksamkeits- und Sicherheitsdaten aus einer klinischen Phase-III-Studie München (19. Oktober 2018) – Vor wenigen Wochen hat die Europäische Kommission die
WEITERLESEN »

Zulassung des ersten RNAi-Therapeutikums: Onpattro® (Patisiran) gibt neue Hoffnung bei hATTR-Amyloidose
Berlin (18. Oktober 2018) – RNAi-Therapeutika leiten eine neue Ära ein, denn sie ermöglichen bei Erbkrankheiten den ursächlichen Fehler in den Genen „stillzulegen“ ohne das Erbgut zu verändern. Mechanismus und Technologie der RNA-Interferenz (RNAi) wurden im Rahmen der Zulassungs-Pressekonferenz von Onpattro® (Patisiran) erörtert, einem der ersten Medikamente dieser Klasse. Onpattro® wird zur Behandlung der
WEITERLESEN »

Hyrimoz® – das Adalimumab-Biosimilar erweitert ab sofort die Therapieoptionen in Rheumatologie, Dermatologie und Gastroenterologie
Biosimilar Hyrimoz® (Adalimumab) für alle Indikationen des Referenzpräparates* zugelassen, einschließlich der Bereiche Rheumatologie, Dermatologie und Gastroenterologie1 Phase-III-Studie ADACCESS2 mit innovativem 4-fach-Crossover-Design belegt, dass der Switch zwischen Original und Biosimilar keinen klinisch relevanten Einfluss hat Der Hyrimoz® SensoReady® Fertigpen bietet eine einfache und patientenorientierte Handhabung für Unabhängigkeit und Sicherheit für den Patienten München (17.
WEITERLESEN »
Erweiterung des Onkologie-Portfolios: Übernahme des Onkologie-Geschäftes von Shire eröffnet neue Perspektiven für Servier
München, Deutschland / Wien, Österreich (29. September 2018) – Am 1. September 2018 übernahm das internationale, private Pharmaunternehmen Servier das Onkologie-Geschäft des irischen Biotechnologie-Unternehmens Shire. Die Transaktion beinhaltet nicht nur zwei bereits verfügbare Onkologie-/ Hämatologie-Produkte, sondern auch Immuno-onkologische Forschungsprojekte. Bei einer Fachpressekonferenz im Rahmen der DGHO-Jahrestagung Ende September in Wien erläuterte Peter Lautenschläger, Business
WEITERLESEN »
Dialog schafft Erkenntnisse – Erkenntnisse schaffen Fortschritt
Pharma-Dialog fortsetzen – Gesundheitswirtschaft verstehen – Rahmenbedingungen definieren Bayer Vital mit guter Position in Deutschland Patientenerfahrung nutzen – Bedürfnisse kennen Präzisionsonkologie: Neue Therapiechancen für Patienten Innovationen: ökonomisch und gesellschaftlich und wichtig Leverkusen (20. September 2018) – Mit Nachdruck spricht sich Frank Schöning für die Fortsetzung des Pharma-Dialogs der Bundesregierung aus. „In einem rohstoffarmen
WEITERLESEN »
Gilead erhält EU-weite Zulassung für Biktarvy® (Bictegravir, Emtricitabin, Tenofoviralafenamid) zur Behandlung der HIV-1-Infektion
In klinischen Studien zeigte Biktarvy eine hohe Wirksamkeit und null Resistenzen über 48 Wochen Martinsried (26. Juni 2018) – Gilead Sciences gab heute bekannt, dass die Europäische Kommission die Marktzulassung für Biktarvy® erteilt hat (Bictegravir 50 mg, Emtricitabin 200 mg, Tenofoviralafenamid 25 mg; BIC/FTC/TAF), ein einmal täglich anzuwendendes Single-Tablet-Regime (STR) für die Behandlung der HIV-1-Infektion. BIC/FTC/TAF
WEITERLESEN »
Europäische Kommission erteilt Zulassungserweiterung für NPLATE® (Romiplostim)
Zulassung basiert auf den Ergebnissen von fünf klinischen Studien Romiplostim-Behandlung reduzierte die Blutungsereignisse bei Kindern mit seltener Bluterkrankung München (9. Februar 2018) – Amgen hat am 1. Februar 2018 bekannt gegeben, dass die Europäische Kommission (EC) die Zulassungserweiterung für Nplate® (Romiplostim) zur Behandlung von Kindern ab einem Jahr mit immun-(idiopathischer)thrombozytopenischer Purpura (ITP) erteilt hat, die
WEITERLESEN »
Mysimba® (Naltrexon/Bupropion)
Greifswald/Frankfurt am Main (31. Januar 2018) – Mysimba®, das weltweit bereits 2 Mio. Mal verordnet wurde, ist ab sofort auch in Deutschland und Österreich verfügbar. Das Präparat ist zur oralen Pharmakotherapie bei Adipositas (BMI ≥ 30 kg/m2) oder Übergewicht (BMI ≥ 27 kg/m2 bei mindestens einer bestehenden gewichtsbezogenen Komorbidität) zugelassen.1 Mehr als die Hälfte der
WEITERLESEN »
Jahrestagung der Deutschen Transplantationsgesellschaft: Tacrolimus in der retardierten Formulierung (MeltDose®-Technologie) zur einmal täglichen Einnahme führt zu flacheren, stabilen Wirkstoffspiegeln
Hamburg (4. Dezember 2017) – Die immunsuppressive Therapie nach Organtransplantation mit Tacrolimus ist heute klinischer Standard. Envarsus® ist ein Tacrolimus-Präparat, das seinen Wirkstoff durch die spezielle MeltDose®-Technologie verzögert freisetzt und so zu flacheren, stabilen Wirkstoffspiegeln im Blut führt. Dadurch können Toxizitätsrisiken durch zu hohe Wirkstoffspiegel reduziert und das Transplantatüberleben durch Vermeidung von Abstoßungen unter zu
WEITERLESEN »
Incyte Biosciences vergrößert klinischen Fußabdruck in Deutschland
Zur Unterstützung seines breiten klinischen Portfolios plant Incyte bei seinen Entwicklungsaktivitäten den Einschluss von mehr als 20 klinischen Prüfungen in Deutschland Planegg/Martinsried (8. November 2017 – Incyte Corporation (NASDAQ: INCY), ein globales biopharmazeutisches Unternehmen, das sich auf die Entdeckung, Entwicklung und Vermarktung neuartiger Arzneimittel zur Deckung dringender medizinischer Versorgungslücken bei Krebs- und anderen Erkrankungen konzentriert,
WEITERLESEN »
Add-on-Therapie der Karzinoid-Syndrom-bedingten Diarrhö: Ipsen Pharma führt Xermelo® (Telotristatethyl) ein
Ettlingen/Frankfurt am Main (18. Oktober 2017) – Seit dem 16.10.2017 steht in Deutschland mit Xermelo (Telotristatethyl) 250 mg eine Add-onTherapie zu Somatostatin-Analoga (SSA) zur Verfügung, die in klinischen Studien(1-2) eine nachhaltige Verbesserung der Karzinoid-Syndrom-bedingten Diarrhö gezeigt hat. Ab dem 01.11.2017 wird der Wirkstoff unter demselben Warenzeichen auch in Österreich verfügbar sein. Im Rahmen der Fachpressekonferenz
WEITERLESEN »
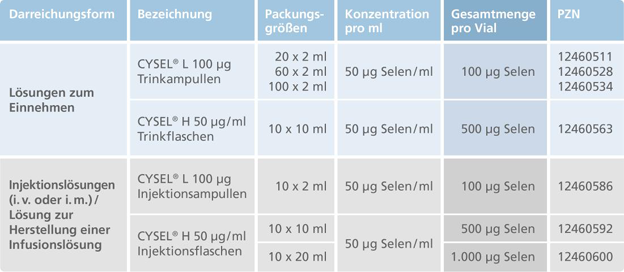
Mundipharma: Neueinführung von Cysel® (Natriumselenit-Pentahydrat) zur Behebung eines nachgewiesenen Selenmangels bei Tumorpatienten
Limburg (30. September 2017) – Mundipharma Deutschland GmbH & Co KG gibt bekannt, dass es am 1. Oktober sein onkologisches Supportivportfolio um das anorganische Selenpräprat Cysel® (Natriumselenit-Pentahydrat) erweitert. Cysel® ist angezeigt zur Behandlung eines klinisch nachgewiesenen Selenmangels, der ernährungsmäßig nicht behoben werden kann.1,2 Cysel® gleicht Selenmangel effektiv aus,1 steigert die Selenoprotein-Aktivität unter oxidativem Stress3,4,5 und
WEITERLESEN »
Therapiedurchbruch bei Riesenzellarteriitis: RoACTEMRA zur Behandlung von Riesenzellarteriitis zugelassen
Grenzach-Wyhlen/Frankfurt am Main (21. September 2017) – Der IL-6-Rezeptorinhibitor RoACTEMRA® (Tocilizumab) ist jetzt auch zur Therapie der Riesenzellarteriitis (RZA) in der EU zugelassen.1 Die Zulassung basiert auf Ergebnissen der Phase-III-Studie GiACTA, der größten klinischen Studie, die bisher bei RZA durchgeführt wurde. Darin erwies sich RoACTEMRA als überlegen wirksam im Vergleich zu Glukokortikoiden (GC), die bislang
WEITERLESEN »
Neuer NK1-Rezeptorantagonist eingeführt: Rolapitant ermöglicht einfache und langanhaltend wirksame Antiemese
München (24. Mai 2017) – Ende April wurde von der EMA mit Rolapitant (VARUBY®) ein neuer Neurokinin-1-Rezeptorantagonist zur Prävention von verzögerter Übelkeit und Erbrechen bei hoch und moderat emetogener Chemotherapie bei erwachsenen Tumorpatienten in der Kombination mit anderen Antiemetika zugelassen. Der moderne Neurokinin-1-Rezeptorantagonist verfügt mit etwa 180 Stunden über eine außerordentlich lange Halbwertszeit. Rolapitant (2
WEITERLESEN »
Roche: Innovationen für Patienten
Penzberg (3. Mai 2017) – Roche zielt darauf, mit innovativen Lösungen neue Therapiestandards für Patienten zu schaffen. Beispielhaft für solche Durchbrüche in der Behandlung von Brust- und Blutkrebs stehen die Original-Biologika Herceptin® (Trastuzumab) und MabThera® (Rituximab). Im Rahmen eines Pressegesprächs am Produktions- und Entwicklungsstandort in Penzberg diskutierten Prof. Tjoung-Won Park-Simon, Hannover, und Prof. Martin Dreyling,
WEITERLESEN »
Von Sandoz beantragte Biosimilars Etanercept und Rituximab zur Zulassung in Europa* empfohlen
Sandoz erhält ein positives CHMP-Gutachten für die Biosimilars Etanercept und Rituximab zur Behandlung von immunologischen Erkrankungen, für das Biosimilar Rituximab auch zur Behandlung von hämatologischen Krebserkrankungen Grundlage für die CHMP-Entscheidungen waren umfassende Datenpakete, mit denen bestätigt wurde, dass die Biosimilars Rituximab und Etanercept ihren jeweiligen Referenzprodukten gleichwertig sind Sofern die Genehmigung der Europäischen Kommission erteilt
WEITERLESEN »
Erste US-Zulassung einer neuen chemischen Substanz (New Chemical Entity, NCE) für Parkinson-Patienten mit motorischen Fluktuationen seit mehr als zehn Jahren: FDA erteilt Marktzulassung für Xadago(R) (Safinamide) für Parkinson-Patienten in den USA
Mailand, Italien und Morristown, NJ, USA (21. März 2017) – Newron Pharmaceuticals S.p.A. ("Newron", SIX: NWRN), ein biopharmazeutisches Unternehmen, das sich auf die Entwicklung neuartiger Therapien für Patienten mit Erkrankungen des zentralen Nervensystems (ZNS) und Schmerzen konzentriert, und seine Partner Zambon S.p.A. und US WorldMeds, LLC, gaben heute die Zulassung von Xadago(R) (Safinamide) durch die
WEITERLESEN »
SGLT2-Inhibitoren (vormals Canagliflozin): Möglicherweise erhöhtes Risiko für Amputationen der unteren Extremitäten
Bonn (10. Februar 2017) – Der PRAC schlussfolgert, dass das Diabetesmittel Canagliflozin zum Risiko von Zehenamputation beitragen kann. Ein Risiko könnte auch für andere Arzneimittel derselben Klasse bestehen. Der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) bei der Europäischen Arzneimittel-Agentur (EMA) warnt, dass in zwei klinischen Prüfungen, CANVAS und CANVAS-R, eine größere Anzahl von
WEITERLESEN »
Gemeinsamer Bundesausschuss veröffentlicht die Nutzenbewertung für Galafold® (Migalastat) bei Morbus Fabry: Mit Migalastat hat erstmals seit Inkrafttreten des AMNOG eine Therapie zur Behandlung der Fabry-Krankheit die frühe Nutzenbewertung durchlaufen
München (1. Dezember 2016) – Amicus Therapeutics (Nasdaq: FOLD), ein führendes Biotechnologie-Unternehmen im Bereich der seltenen Erkrankungen, gab heute bekannt, dass der Gemeinsame Bundesausschuss (G-BA) die Bewertung des Zusatznutzens von Galafold® (Migalastat) abgeschlossen und veröffentlicht hat. Der Zusatznutzen des oral einzusetzenden Chaperons Migalastat wurde für die Langzeitbehandlung von Erwachsenen und Jugendlichen ab 16 Jahren mit
WEITERLESEN »
Rote-Hand-Brief zu levetiracetamhaltigen Lösungen zum Einnehmen: Risiko einer Überdosierung durch Medikationsfehler
Bonn (21. November 2016) – Bei der Verabreichung von Levetiracetamlösung (100 mg/ml) zum Einnehmen war es in mehreren Fällen zu einer versehentlichen, bis zu zehnfachen Überdosierung, insbesondere bei Kindern im Alter bis zu elf Jahren gekommen. Als wichtige Ursache wurde die Verwendung einer falschen Dosiervorrichtung identifiziert. Überdosierung von Levetiracetam kann zu herabgesetztem Bewusstsein, Atemdepression und
WEITERLESEN »
Rote-Hand-Brief zu Revlimid® (Lenalidomid): Neuer wichtiger Hinweis zur Reaktivierung von Virusinfektionen
Bonn (8. November 2016) – Die Firma Celgene GmbH informiert darüber, dass Fälle von Virus-Reaktivierung nach der Behandlung mit Lenalidomid berichtet wurden, insbesondere bei Patienten, die zuvor mit Herpes-Zoster- oder Hepatitis-B-Viren (HBV) infiziert worden waren. Bei einigen Fällen führte die HBV-Reaktivierung zu einem akuten Leberversagen und zum Tod. Der HBV-Status ist vor Beginn der
WEITERLESEN »