


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Pathophysiologie der PAH im Kindesalter
Prof. Dr. med. J. Breuer
Weimar (4. Oktober 2009) – Pulmonal arterielle Hypertonie im Kindesalter ist eine sehr seltene, schwerwiegende Erkrankung. Bleibt sie nach der Diagnosestellung unbehandelt, ist die geschätzte mittlere Überlebensdauer nur ca. 10 Monate. (Widlitz A et Barst RJ 2003). Eine pulmonal arterielle Hypertonie kann im Kindes- und Neugeborenenalter auf sehr verschiedenen Grunderkrankungen beruhen. Ursachen sind hauptsächlich die persistierende pulmonal Hypertonie des Neugeborenen (PPHN), die bronchopulmonale Dysplasie (BPD), die pulmonale Hypertonie bei angeborenen Herzfehlern, die idiopathische pulmonal arterielle Hypertonie (IPAH) oder auch das hypoxische Lungenversagen.
Bei etwa 90 % der betroffenen Kinder liegt entweder eine idiopathische PAH (IPAH), hereditäre PAH (HPAH, früher familiäre PAH, FPAH) oder eine PAH im Zusammenhang mit einem angeborenen Herzfehler (APAH-CHD) vor (Simonneau G et al. 2009).
Das Lungengefäßsystem reagiert offensichtlich in gleicher Weise auf verschiedene Auslöser mit einer pulmonalen Hypertonie. Dies wird auch in der zuletzt in Dana Point 2008 aktualisierten klinischen Klassifikation der pulmonalen Hypertonie deutlich.
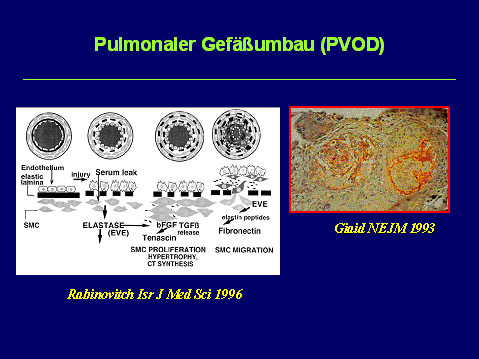
Im Zentrum der Pathogenese steht eine Dysfunktion des pulmonalen Endothels, die sowohl zu einer Störung der Balance zwischen vasodilatatorischen (NO, Prostazyklin) und vasokonstriktorischen Faktoren (Endothelin, Thromboxan, Angiotensin II) als auch zu einer Zellproliferation, einer gestörten Apoptose sowie einer veränderten Wechselwirkung von Endothel- und glatten Muskelzellen führen kann. Eine inflammatorische Gefäßwandschädigung mit konsekutiver Thrombozytenaggregation führt zu einer weiteren Störung der Lungenperfusion. Die endotheliale Dysfunktion beruht bei der IPAH und HPAH unter anderem auf einem Defekt im sogenannten TGF-ß-Signalweg (transforming growth factor ß).
In verschiedenen Tiermodellen mit systemisch-pulmonalen Shunts konnte eine Überexpression von ET-1 und der entsprechenden Rezeptoren nachgewiesen werden. Auch im Plasma von Kindern mit angeborenen Herzfehlern und bei Patienten mit erhöhtem pulmonalen Blutfluss oder Links-Rechts-Shunt wurden erhöhte ET-1-Werte gefunden (Beghetti M et al. 2005). Durch die Blockierung der ET-Rezeptoren kann die Entwicklung einer PAH im Tiermodell verhindert werden, auch scheint ET-1 bereits zu einem sehr frühen Zeitpunkt der pulmonal-vaskulären Veränderungen eine Rolle zu spielen (Rondelet B et al. 2003). Insgesamt sprechen sowohl tierexperimentelle als auch klinische Daten dafür, dass ET-1 wesentlich zur Entwicklung einer PAH Kindern sowohl mit der hereditären PAH wie auch der mit angeborenen Herzfehlern assoziierten Form beiträgt.
Bosentan – ein dualer Endothelin-Rezeptor-Antagonist, kann durch die Blockade der Endothelin-Rezeptoren ETA und ETB sowohl die Vasokonstriktion, als auch Inflammation und Fibrosierung der Gefäße positiv beeinflussen und dadurch die Rechtsherzhypertrophie günstig beeinflussen.
Liegt ein angeborener Herzfehler, z. B. ein Shunt-Vitium mit einem ausgeprägten links-rechts-Shunt, vor, spielen vermehrte Scherkräfte am Gefäßendothel eine wichtige Rolle. Eine progrediente Zunahme des pulmonalen Gefäßwiderstandes führt bei Kindern mit einem solchen Shunt-Vitium schließlich zu einer Shunt-Umkehr, die als Eisenmengerreaktion bezeichnet wird. Diese Patienten haben, unter anderem aufgrund der durch das Shunt-Vitium bestehenden Entlastung des rechten Ventrikels, unbehandelt eine etwas bessere Prognose als die Patienten mit einer IPAH oder HPAH.
Eine pathophysiologisch orientierte Therapie sollte neben einer möglichen Beseitigung der zugrunde liegenden Ursache nicht nur auf eine Vasodilatation, sondern auch auf eine Beeinflussung der gesteigerten Proliferation und Inflammation im pulmonalen Gefäßsystem gerichtet sein.
Kernaussagen
-
Pulmonal arterielle Hypertonie im Kindesalter ist eine sehr seltene, schwerwiegende Erkrankung.
-
Eine progressive Vaskulopathie der kleinen pulmonalen Arterien kann bis zum Rechtsherzversagen führen. Bleibt die pädiatrische PAH nach der Diagnosestellung unbehandelt, ist die geschätzte mittlere Überlebensdauer nur ca. 10 Monate.
-
Die endotheliale Dysfunktion der pulmonalen Arterien führt zu einer reduzierten Vasodilatation und durch die erhöhte Freisetzung von gefäßverengenden Mediatoren, wie z. B. Endothelin, einer vermehrten Vasokonstriktion.
-
Eine pathophysiologisch orientierte Therapie sollte neben einer möglichen Beseitigung der zugrunde liegenden Ursache nicht nur auf eine Vasodilatation, sondern auch auf eine Beeinflussung der gesteigerten Proliferation und Inflammation im pulmonalen Gefäßsystem gerichtet sein.
Literatur
-
Beghetti M et al. Endothelin-1 in congenital heart disease. Ped Res 2005;57:16R-20R.
-
Rondelet B et al. Bosentan for the prevention of overcirculation-induced experimental pulmonary arterial hypertension. Circulation 2003; 107: 1329-1335.
-
Schulze-Neick I. Pulmonal arterielle Hypertonie bei angeborenen Herzfehlern. Dtsch Med Wochenschr 2006; 131: S322-S324.
-
Simonneau G et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009; 54 (1 Suppl): S43-54.
-
Schranz D. Pulmonale Hypertonie im Kindesalter. Intensivmedizin up2date 2 2006; 2: 177-193.
-
Widlitz A et Barst RJ. Pulmonary arterial hypertension in children Eur Respir J 2003; 21: 155–176.
Abbildungen

Abb. 1: Pulmonaler Gefäßumbau (PVOD).

Abb. 2: ET-Rezeptoren unter physiologischen und unter pathologischen Bedingungen.

Abb. 3: PAH und CHD.

Abb. 4: Entwicklung eines Eisenmenger-Syndroms.
Quelle: Actelion-Symposium zum Thema „Therapie von Anfang an – pulmonal arterielle Hypertonie (PAH) bei Kindern und Jugendlichen“ am 04.10.2009 in Weimar, anlässlich des Kongresses der Deutschen Gesellschaft für pädiatrische Kardiologie (DGPK) (Cramer-Gesundheits-Consulting) (tB).