


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Therapieziele erreichen – praktisches Management der PAH nach aktuellen Leitlinien
Prof. Dr. med. Marius M. Hoeper
Hannover (18. März 2010) – Die 2009 gemeinsam von der European Society of Cardiology (ESC) und der European Respiratory Society (ERS) herausgegebenen Leitlinien zur Diagnostik und Therapie der pulmonalen Hypertonie unterscheiden sich in zentralen Punkten von älteren Leitlinien.1, 2 Die vielleicht wichtigste Neuerung ist die Formulierung von Therapiezielen für Patienten mit pulmonal arterieller Hypertonie (PAH). Entscheidend ist die Festlegung solcher Therapieziele für die Steuerung der Therapie, insbesondere für die Entscheidung, ob und wann eine Kombinationstherapie erforderlich wird. Die in den Leitlinien vorgegebenen Therapieziele basieren auf Parametern mit gut dokumentierter prognostischer Bedeutung bei Patienten mit PAH. Diese Parameter lassen sich in 3 Gruppen unterteilen:
-
Zeichen der klinischen Instabilität (Symptome der Rechtsherzinsuffizienz, Krankheitsprogression, Synkopen),
-
Parameter der körperlichen Belastbarkeit (Verbesserung und Stabilisierung der WHO-/NYHA-Klasse, 6-Minuten-Gehstrecke, Spiroergometrie), sowie
-
Parameter der Rechtsherzfunktion (BNP bzw. NT-proBNP, Rechtsherzfunktion in der Echokardiographie sowie in der invasiven Diagnostik).
Es ist nicht erforderlich, sämtliche Parameter bei jeder Patientenvorstellung zu überprüfen. Primär muss sichergestellt werden, dass die Erkrankung nicht progredient ist. Bei klinisch stabilen Patienten mit befriedigendem Therapieergebnis genügen in der Regel die Durchführung eines Belastungstests sowie die Evaluation der Rechtsherzfunktion mit einem der genannten Verfahren.
Die Höhe des pulmonal-arteriellen Drucks, unabhängig davon, ob echokardiographisch abgeschätzt oder invasiv ermittelt, spielt für die prognostische Beurteilung von Patienten mit PAH und somit für therapeutische Entscheidungen praktisch keine Rolle.3, 4
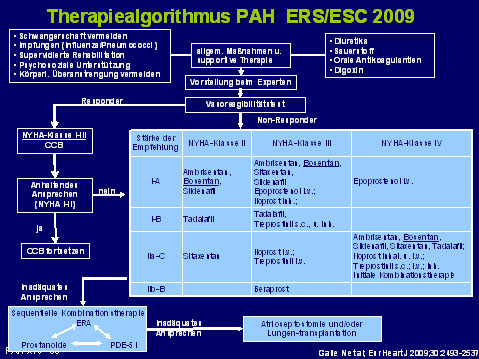
Bei PAH im funktionellen Stadium NYHA II oder III empfehlen die aktuellen Leitlinien zunächst eine Monotherapie. In der Regel werden in diesen Krankheitsstadien Endothelin-Rezeptor-Antagonisten oder Phosphodiesterase-5 (PDE-5) Inhibitoren eingesetzt.5, 6
Je nach klinischer Situation sollte im weiteren Verlauf in etwa 3-monatigen Abständen überprüft werden, ob die Therapieziele erreicht werden. Sollte dies nicht der Fall sein, wird eine Therapieerweiterung im Sinne einer Kombinationstherapie empfohlen. Bei PAH-Patienten, die sich in der WHO-/NYHA-Klasse IV vorstellen, kann auch primär eine Kombinationstherapie erwogen werden. Dieses Vorgehen hat sich in der klinischen Praxis bereits weitgehend durchgesetzt, wobei die in den Leitlinien genannten Therapieziele der individuellen Situation des Patienten angepasst werden müssen.
FAZIT
Die aktuellen Leitlinien zur Diagnostik und Therapie der pulmonalen Hypertonie legen zum ersten Mal Therapieziele wie zum Beispiel Besserung und Stabilisierung der WHO-/NYHA-Klasse mindestens auf WHO-/NYHA-Klasse II oder Besserung der Hämodynamik für Patienten mit pulmonal arterieller Hypertonie (PAH) zur Steuerung der Therapie fest.
In etwa 3-monatigen Abständen soll die Zielerreichung überprüft werden. Sollten die Therapieziele nicht erreicht werden, wird eine Therapieerweiterung im Sinne einer Kombinationstherapie empfohlen.
Literatur
-
Galiè N et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. The task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Respir J 2009; 34:1219-1263.
-
Galiè N et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493-2537.
-
Sitbon O et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40:780-788.
-
McLaughlin VV et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005;25:244-249.
-
Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J 2008;31:407-414.
-
Wilkins MRet al. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Respir J 2008;32:198-209.
Abbildungen

Abb. 1: Therapiealgorithmus PAH ERS/ESC 2009.

Abb. 2: Empfohlene Verlaufsuntersuchungen bei Patienten mit PAH.

Abb. 3: Prognostische Parameter bei pulmonal arterieller Hypertonie.
Quelle: Satelliten-Symposium der Firma Actelion zum Thema „Pulmonal arterielle Hypertonie und darüber hinaus – das Therapieziel zählt“ am 18.03.2010 in Hannover , anlässlich der Jahrestagung der Deutschen Gesellschaft für Pneumologie (Cramer-Gesundheits-Consulting) (tB).