


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Lyosomale Speicherkrankheiten
Neue Therapieoptionen: Falldemonstrationen
Von Prof. Dr. med. Michael Beck, Mainz
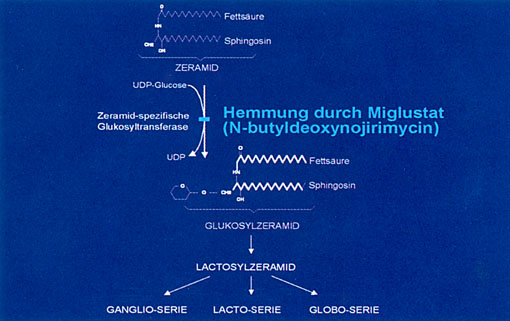
Wiesbaden (16. April 2007) – Die Zeramid‑spezifische Glykosyltransferase synthetisiert Glukosylzeramid aus Zeramid und UDP‑Glukose. Sie stellt ein Schlüsselenzym für die Synthese von komplexen Lipiden (Glukosylzeramid, Gangliosiden, Globosiden u.a.) dar, die sich bei verschiedenen Lipidspeicherkrankeiten (M. Gaucher, M. NiemannPick) anreichern.
Bahnbrechende Arbeiten haben gezeigt, dass das Enzym Glykosyltransferase durch den Iminozucker N‑butyldeoxynojirimycin (Miglustat) reversibel gehemmt werden kann (Platt FM et al 1994). Aufgrund dieser Ergebnisse wurden mehrere klinische Studien mit Miglustat bei Patienten mit M. Gaucher durchgeführt, die die klinische Wirksamkeit belegen konnten. Es kam zu einem Rückgang der Leber‑ und Milzvergrößerung, Normalisierung des Blutbildes und Abfall des Enzyms Chitotriosidase, das als Marker für den Krankheitsverlauf dient (Cox, T. et ah 2000). Langzeitstudien haben gezeigt, dass die Behandlung mit Miglustat auch zu einem Rückgang der Lipidspeichenung im Knochenmark und damit zu einer Stabilisierung des Skelettsystems führt (Eistein, D. et al 2004). Miglustat ist zugelassen zur Behandlung des leichten bis mittelschweren M. Gaucher bei erwachsenen Patienten, bei denen eine Enzymersatztherapie aus medizinischen oder persönlichen Gründen nicht indiziert ist (Cox. T.M. et al 2003). Miglustat ist gut verträglich; Durchfälle und eine (reversible) Gewichtsabnahme gehören zu den häufigsten Nebenwirkungen.
Der M. Niemann‑Pick Typ C stellt ist eine Lipidspeicherkrankheit, die durch einen angeborenen Defekt im intrazellulären Fettstoffwechsel verursacht ist und eine progrediente lysosomale Speicherung von Cholesterin und Glykolipiden bewirkt. Führende Symptome dieser neurodegenerativen Krankheit sind Leber‑ und Milzvergrößerung, vertikale Blickparese, zerebelläre Ataxie, Dysarthrie, Krampfanfälle, Kataplexie und Demenz. Eine ursächliche Therapie stand bisher nicht zur Verfügung.
Im Tierversuch (Mausmodell des M. Niemann‑Pick Typ C) zeigte sich ein Rückgang der Lipidspeicherung durch den Glykolipidsynthese‑Inhibitor Miglustat, auch waren bei den behandelten Tieren die neurologischen Symptome deutlich abgeschwächt (Zervas, M. et al 2001). Aufgrund dieser Ergebnisse wurde eine klinische Studie initiiert, an der 41 Patienten (29 Erwachsene und 12 Kinder unter 12 Jahren) teilnahmen. Nach einer Behandlung mit Miglustat über ein Jahr konnte ein positiver Effekt auf den Krankheitsverlauf festgestellt werden (Patterson, M.C. et al 2006): Die Augenmotilitätsstörung besserte sich, Schluckbeschwerden gingen zurück, weiterhin wurde eine Besserung der kognitiven Fähigkeiten beobachtet.
Fallbericht eines Geschwisterpaares mit M. Niemann‑Pick Typ C*: Die Diagnose wurde bei der älteren der beiden Schwestern mit 5 1/2 Jahren aufgrund einer vertikalen Blickparese und motorischer Ungeschicklichkeit gestellt. Im folgenden kam es zu einer rasch fortschreitenden Verschlechterung mit Rollstuhlpflichtigkeit, autistischem Erscheinungsbild und therapieresistenter Epilepsie im Alter von 7 Jahren; das Mädchen verstarb im Alter von 9 1/2 Jahren. Die 2 Jahre jüngere Schwester zeigte im Alter von 51/2 Jahren ebenfalls eine vertikale Blickparese. Nach Beginn einer Therapie mit Miglustat konnten die motorischen Fähigkeiten bei weiter bestehender vertikaler Blickparese über einen Zeitraum von 2 Jahren weitestgehend stabilisiert werden. Auch die kognitive Leistungsfähigkeit, die in jährlichen Kontrolluntersuchungen beurteilt wurde, blieb stabil. Die akustisch evozierten Potentiale (AEP) und die somatosensorisch evozierten Potentiale (SSEP) waren stets unauffällig. Die visuell evozierten Potentiale (VEP) zeigten zu Therapiebeginn eine hohe kortikale Antwort, die im weiteren Verlauf nach 2 Jahren eine abnehmende Tendenz zeigte. Der Vergleich der beiden Geschwister zeigt eindrucksvoll den unterschiedlichen Verlauf der Erkrankung mit versus ohne Therapie. In Anbetracht der zukünftig möglichen Therapie ist es besonders wichtig, die Zeit vom Beginn der ersten Symptome bis zur Diagnose zu verkürzen, um die Behandlung dann so früh wie möglich einleiten zu können.
Zusammenfassend stellt Miglustat schon heute eine wirksame Therapieoption für Patienten mit leichtem bis mittelschwerem M. Gaucher dar; die Substratregulationstherapie führt zu einer Reduktion der Hepatosplenomegalie, Verbesserung hämatologischer und biochemischer Parameter und einer Zunahme der Knochenmarkfettfraktion mit Rückgang von Knochenschmerzen und Stabilisierung des Skelettsystems.
Anmerkung
* Zavesca® (Miglustat ist in dieser Indikation nicht zugelassen.
Keyslides des Vortrages:

Abb. 1: Hemmung durch Miglustat.

Abb. 2: M. Gaucher: Veränderung der Fettfraktion des Knochenmarks unter Miglustat.

Abb.: Geschwisterpaar mit M. Niemann-Pick Typ C: Vergleich klinischer Daten.

Abb. 4: Substratregulationstherapie im Überblick.
Referenzen
-
Platt, F.M et al; J Biol Chem 1994;2698362-8365
-
Cox, T. et al; Lancet 2000,355.1481-1485
-
Elstein, 0 et al; J Inherit Metab Dis 2004;27.757-766
-
Cox, T.M. et all J Inherit Metab Dis 2003;26.513-526
-
Zervas, M. et all Curr Biol 2001;11:1283-1287
-
Patterson, M.C. et all American Society of Human Genetics (ASGH) 2006,425
Quelle: Satelliten-Symposium der Firma Actelion Pharmaceuticals Deutschland zum Thema “Lyosomale Speicherkrankheiten im Fokus – viele Gesichter einer Gruppe seltener Erkrankungen“ anlässlich des 113. Kongresses der Deutschen Gesellschaft für Innere Medizin (DEGIM) am 16.04.2007 in Wiesbaden (CGC – Cramer-Gesundheits-Consulting).