


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Pulmonale Hypertonie bei interstitiellen Lungenerkrankungen (ILE) – therapeutische Möglichkeiten
PD Dr. med. Jürgen Behr
Hannover (18. März 2010) – Interstitielle Lungenerkrankungen sind charakterisiert durch einen bindegewebigen Umbau des Alveolargewebes. Inflammation und Fibroproliferation sind – je nach Grunderkrankung – in unterschiedlichem Ausmaß an den Umbauprozessen beteiligt. Die Lungenfunktion wird bei interstitieller Lungenerkrankung von einer restriktiven Ventilationsstörung sowie einer Einschränkung der Diffusionskapazität dominiert. Die Beteiligung der Lungengefäße in diesem Krankheitsprozess bedingt die Entwicklung einer präkapillären pulmonalen Hypertonie. Beispiel hierfür ist die idiopathische Lungenfibrose (IPF), bei der sich Intimaläsionen zu avaskulären Fibrosearealen mit vollständiger Obliteration des ursprünglichen Gefäßlumens entwickeln können. Auch bei der Sarkoidose ist ein direkter Gefäßbefall mit Obliteration des Lumens beschrieben.
Die Prävalenz der pulmonalen Hypertonie (PH) bei interstitiellen Lungenerkrankungen variiert stark in Abhängigkeit von Patientenselektion und angewandter Untersuchungstechnik: Sklerodermie 5‑38 %, systemischer Lupus Erythematodes 4‑43 %, rheumatoide Arthritis ca. 20 % und Sarkoidose 1‑28 %. Das Bestehen einer zusätzlichen PH macht sich oft durch eine im Vergleich zur restriktiven Funktionseinschränkung überproportionale Verminderung der Diffusionskapazität, des O2-Bedarfs und der physischen Leistungsfähigkeit bemerkbar. Zahlreiche Untersuchungen belegen, dass das Auftreten einer PH im Rahmen einer interstitiellen Lungenerkrankung immer mit einer ungünstigen Prognose assoziiert ist.
Hinsichtlich der pathophysiologischen Rolle der PH stellt sich die Frage, ob das Auftreten einer PH als Zeichen eines fortgeschrittenen Krankheitsstadiums anzusehen ist, das ´per se` eine ungünstigere Prognose mit sich bringt – oder ob Patienten, die im Rahmen einer interstitiellen Lungenerkrankung eine PH entwickeln, möglicherweise eine prognostisch belastete Untergruppe darstellen, bei der die pulmonale Hypertonie als solche den Krankheitsverlauf und auch die Belastbarkeit der Patienten determiniert. Im ersteren Falle wäre eine spezifische Therapie der PH voraussichtlich wenig wirkungsvoll, da die schwere restriktive Lungenerkrankung dominiert. Im zweiten Fall dagegen könnte eine spezifische Behandlung der PH den Krankheitsverlauf und die Belastbarkeit der Patienten günstig beeinflussen.
Die Therapie der mit interstitiellen Lungenerkrankungen assoziierten pulmonalen Hypertonie sollte mit einer optimalen Behandlung der interstitiellen Grunderkrankung beginnen. In Hinblick auf mögliche therapeutische Interventionen liegen jedoch nur rudimentäre Daten in der Literatur vor. Für die häufig bei unterschiedlichen interstitiellen Lungenerkrankungen durchgeführte immunsuppressive Therapie mit Prednisolon und Kombinationen mit Zytostatika gibt es, abgesehen von einigen Fallberichten, keine hinreichenden Belege für eine Wirksamkeit in Hinblick auf die PH.
In mehreren Akutstudien konnte gezeigt werden, dass die zur Verfügung stehenden Substanzen zur Senkung des pulmonalen arteriellen Druckes – NO inhalativ, Epoprostenol oder Iloprost i.v. oder inhalativ, Sildenafil, etc. – diese Wirkung auch bei Patienten mit Lungenfibrose und pulmonaler Hypertonie hervorrufen.
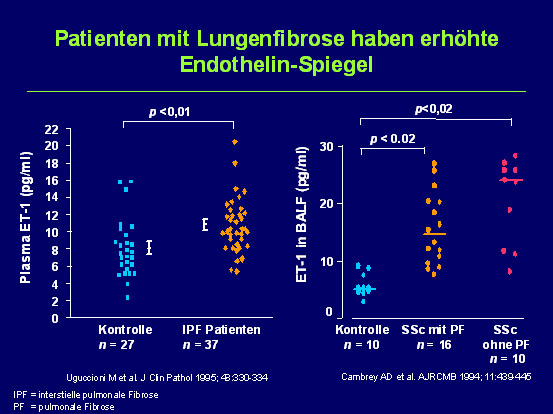
Antiproliferative Wirkungskomponenten der hierbei eingesetzten Substanzen könnten sich zusätzlich positiv auf den fibroproliferativen Prozess bei interstitiellen Lungenerkrankungen auswirken. Dies gilt in besonderer Weise für Endothelin-Rezeptor-Antagonisten, weil Endothelin-1 einerseits eine ausgeprägte proliferative Wirkung aufweist und andererseits im Serum von Patienten mit Lungenfibrose ohne und mit assoziierter pulmonaler Hypertonie in erhöhter Konzentration nachgewiesen wurde. Dementsprechend wird die Wirkung von Endothelin-Rezeptor-Antagonisten auf die IPF selbst (BUILD-1-, BUILD-3-, MUSIC- und ARTEMIS-IPF-Studie) aber auch auf die IPF-assozierte PH (Artemis-PH-Studie) derzeit in prospektiven, randomisierten Doppelblind-Studien untersucht. Die BUILD-1-Studie hat für Bosentan zumindest bei wenig fortgeschrittenem Lungenparenchymumbau einen günstigen Effekt auf die Krankheitsprogression gezeigt.
Entsprechend der weitgehend fehlenden Datenlage empfehlen die WHO-Konferenz von Dana Point 2008 und die ERS/ESC-Leitlinien zur PH 2009 übereinstimmend eine spezifische PH-Therapie bei Patienten mit interstitiellen Lungenerkrankungen nur im Rahmen von klinischen Studien und die Vorstellung entsprechender Patienten in PH-Zentren, die einerseits Studien durchführen und andererseits geeignete Patienten auch einer Lungentransplantation als definitives Therapieverfahren zuführen können.
FAZIT
-
Interstitielle Lungenerkrankungen sind charakterisiert durch einen bindegewebigen Umbau des Alveolargewebes, Inflammation und Fibroproliferation und führen bei Beteiligung der kleinen Lungengefäße zur präkapillären pulmonalen Hypertonie.
-
Endothelin-I ist an der Pathogenese der pulmonalen Hypertonie bei interstitiellen Lungenerkrankungen beteiligt.
-
Die optimale Behandlung der zugrunde liegenden Lungenerkrankung einschließlich Langzeit-Sauerstoff-Therapie für Patienten mit chronischer Hypoxie ist Grundlage der Therapie.
-
Die BUILD-1-Studie hat für den dualen Endothelin-Rezeptor-Antagonisten Bosentan bei wenig fortgeschrittenem Lungenparenchymumbau einen günstigen Effekt auf die Krankheitsprogression der pulmonalen Hypertonie gezeigt.
-
Patienten mit pulmonaler Hypertonie auf Grund von Lungenerkrankungen sollten in RCT eingeschlossen werden, die PAH-spezifische Medikamente untersuchen (z. B. BUILD-3).
Literatur
-
King TE et al. BUILD-1: A randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008;177:75-81.
-
Galiè N et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009;34:1219–1263.
-
Hoeper MM et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009; 54(1 Suppl):S85-96.
Abbildungen

Abb. 1: Patienten mit Lungenfibrose haben erhöhte Endothelin-Spiegel.

Abb. 2: BUILD 1: Bosentan verzögert die Zeit bis zur Krankheitsprogression oder Tod bei Patienten mit histologisch gesicherter IPF.

Abb. 3: Leitlinien pulmonale Hypertonie 2009.
Quelle: Satelliten-Symposium der Firma Actelion zum Thema „Pulmonal arterielle Hypertonie und darüber hinaus – das Therapieziel zählt“ am 18.03.2010 in Hannover , anlässlich der Jahrestagung der Deutschen Gesellschaft für Pneumologie (Cramer-Gesundheits-Consulting) (tB).