


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Lysosomale Speichererkrankungen: selten gesehen – oft übersehen
Eine Einführung
Von Prof. Dr. med. Anibh M. Das
München (12. September 2008) – Das klinische Bild lysosomaler Speichererkrankungen ist seit Langem bekannt. So wurden die Symptome des M. Gaucher bereits 1882 von Phillipe Gaucher und das klinische Bild des M. Anderson-Fabry 1898 unabhängig von William Anderson und Johannes Fabry beschrieben. Vor gut 50 Jahren wurden die Lysosomen von der Arbeitsgruppe um Christian de Duve entdeckt, was die biochemische Charakterisierung dieser Erkrankungen als lysosomale Speichererkrankungen möglich machte. Es war Géry Hers, der 1963 als Erster die Pathogenese einer lysosomalen Speichererkrankung beim M. Pompe durch Nachweis einer Defizienz der sauren alpha-Glucosidase aufklären konnte. In den folgenden Jahren wurden mehr und mehr lysosomale Speichererkrankungen biochemisch charakterisiert. Im Verlauf gelang es auch, bei vielen dieser Erkrankungen die molekulargenetische Ursache aufzuklären.
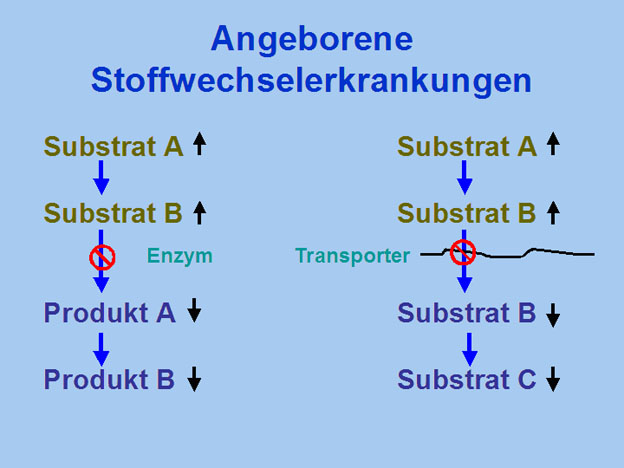
Die lysosomalen Speichererkrankungen zählen gegenwärtig nicht zu den Zielkrankheiten des bei jedem Neugeborenen in den ersten Lebenstagen in Deutschland, Österreich und der Schweiz durchgeführten Screenings auf angeborene Stoffwechselerkrankungen. Gleichwohl ist die kollektive Häufigkeit der lysosomalen Speichererkrankungen hoch und wird auf 1:7700 Lebendgeburten geschätzt. Die Diagnose muss daher klinisch gestellt werden. Da zahlreiche lysosomale Stoffwechselerkrankungen sich bereits im Kindesalter klinisch manifestieren, kommt dem/der Pädiater(in) hierbei eine Schlüsselrolle zu. Der/Die Pädiater(in) sollte mit den typischen Symptomen lysosomaler Speichererkrankungen vertraut sein, damit die selektive Konfirmationsdiagnostik umgehend eingeleitet werden kann.
Im Focus der Entwicklungen steht in letzter Zeit die Etablierung therapeutischer Optionen. Für mehrere lysosomale Speichererkrankungen gelang die biotechnologische Herstellung des defizienten Enzyms, das den Patienten regelmäßig infundiert wird. Beispiele hierfür sind der M. Gaucher, M. Fabry, M. (Hurler-) Scheie, M. Pompe, M. Maroteaux-Lamy (MPS VI) und M. Hunter. Extrazerebrale Symptome können dadurch positiv beeinflusst werden. Aufgrund ihrer Größe können die infundierten Enzyme die Blut-Hirn Schranke jedoch nicht überwinden, so dass der Einfluss auf neurologische Symptome unzureichend ist. Eine weitere Möglichkeit des Enzymersatzes ist die Knochenmarktransplantation, die auch das Hirn erreicht. Insbesondere beim M. Hurler ist diese Therapieform auch hinsichtlich der psychomotorischen Entwicklung Erfolg versprechend. Viel versprechend ist die Substratreduktionstherapie durch kleine Moleküle wie Miglustat (Zavesca®), einer Imino-Zucker Verbindung, die auch ins Gehirn gelangt. Durch diese Substanz wird die Biosynthese von Glycosphingolipiden, die bei lysosomalen Speichererkrankungen nicht abgebaut werden können, gehemmt. Somit fällt weniger Speichermaterial an und es kommt zu einer Entspeicherung. Miglustat (Zavesca®) wirkt auch als Chaperon, indem es die Proteinfaltung defekter lysosomaler Enzyme beeinflusst, wodurch das defekte Enzym stabilisiert und seine Aufnahme in die Lysosomen gefördert wird. Mit dieser Substanz gibt es beim M. Gaucher langjährige Erfahrungen, auch andere Speichererkrankungen sind einer solchen Therapie zugänglich.
Der Vererbungsmodus von lysosomalen Speichererkrankungen ist variabel, einige Erkrankungen werden autosomal-rezessiv, andere X-chromosomal vererbt, was auch für andere angeborene Stoffwechselerkrankungen zutrifft. Diesem Thema widmet sich der erste Vortrag des Symposiums. Im zweiten Vortrag stehen die klinischen Symptome lysosomaler Speichererkrankungen im Vordergrund. Da die Erkrankungen aus diesem Formenkreis nicht zu den Zielkrankheiten des Neugeborenenscreenings gehören, wird die Diagnose primär klinisch gestellt. Der/Die Pädiater(in) sollte daher mit dem klinischen Bild vertraut sein, um die Diagnose zu stellen bzw. eine Konfirmationsdiagnostik zu veranlassen. Der abschließende Vortrag befasst sich mit Knochenmanifestationen bei M. Gaucher.

Abb. 1

Abb. 2

Abb. 3

Abb. 4
Referent
Prof. Dr. med. Anibh M. Das
Pädiatrie II
Medizinische Hochschule Hannover
Carl-Neuberg-Straße 1
30625 Hannover
Quelle: Satelliten-Symposium der Firma Actelion zum Thema „Lyosomale Speichererkrankungen: selten gesehen – oft übersehen“ am 12.09.2008 in München, anlässlich der 104. Jahrestagung der Deutschen Gesellschaft für Kinder- und Jugendmedizin (CGC Cramer-Gesundheits-Consulting) (tB).