


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Stoffwechselkrankheiten – Genetische Grundlagen
Von Prof. Dr. med. Michael Beck
München (12. September 2008) – Lysosomale Speicherkrankheiten stellen eine große Gruppe von Stoffwechselkrankheiten dar; sie beruhen auf einer Funktionsstörung der Lysosomen. Die meisten lysosomalen Speicherkrankheiten werden durch einen Defekt eines Enzyms verursacht; einigen (seltenen) Formen liegen komplexere Störungen zugrunde (z. B. Aktivatordefekt, Transportdefekt usw.). In letzter Zeit wurde eine Erkrankung (Danon Disease) bekannt, die auf einem Defekt der lysosomalen Membran, beruht.
Lysosomale Speicherkrankheiten (z. B. Morbus Gaucher, Morbus Niemann-Pick) folgen zumeist dem autosomal-rezessiven Erbgang.
Die Gaucher-Erkrankung ist die häufigste lysosomale Glykosphingolipidose und durch eine Akkumulation von Glukozerebrosid in den Makrophagen charakterisiert. Klinisch manifestiert sich die nicht-neuropathische Form des M. Gaucher typischerweise mit der Trias Hepatosplenomegalie, Knochenbefall und Laborwertveränderungen. Die akut neuropathische Verlaufsform ist charakterisiert durch schwere neurologische Komplikationen, die in der Regel innerhalb der ersten zwei Lebensjahre zum Tode führen, während der chronisch neuropathische Typ durch leichtere neurologische Symptome und eine geringere Progredienz gekennzeichnet ist.
Niemann-Pick ist eine extrem seltene genetische Störung, die auf einer pathologischen Überladung von Nervenzellen mit Glykosphingolipiden infolge eines gestörten Lipidtransportes in der Zelle beruht. Cholesterin, das aus dem Kreislauf über Low-Density-Lipoprotein- (LDL-) Rezeptoren in die Zelle aufgenommen wird, kann in den parenchymatösen Organen sowie im Zentralnervensystem (ZNS) nicht auf normalem Wege zeitgerecht verarbeitet werden. Lipide reichern sich in toxischen Mengen in Form von nativem Cholesterin, Sphingomyelin, Phospholipiden und Glykolipiden an, wodurch es zu strukturellen und funktionellen Schäden an Zellen und Geweben kommt. Niemann-Pick Typ C (NPC) zeigt, bei einem in der Mehrzahl der Fälle tödlichen Verlauf und ein extrem heterogenes Erscheinungsbild. Die Erkrankung kann, mit oder ohne eine begleitende Hepatosplenomegalie, bei Säuglingen, Kindern oder Erwachsenen auftreten, und ist durch Störungen der Okulomotorik, Dysphagie und Dysarthrie, Ataxie sowie fortschreitende kognitive Störungen, die zu einer Demenz führen, gekennzeichnet. Die Diagnose des M. Niemann-Pick Typ C1 und Typ C2 beruht zunächst auf dem Nachweis einer vermehrten Anreicherung von freiem Cholesterin in Fibroblasten, um die Diagnose sicher zu stellen, ist jedoch eine Gen-Analyse erforderlich.
Bei lysosomalen Speicherkrankheiten besteht keine strenge Genotyp/Phänotyp-Beziehung, im Einzelfall können der Schweregrad und der Verlauf einer Erkrankung nicht vorhergesagt werden.
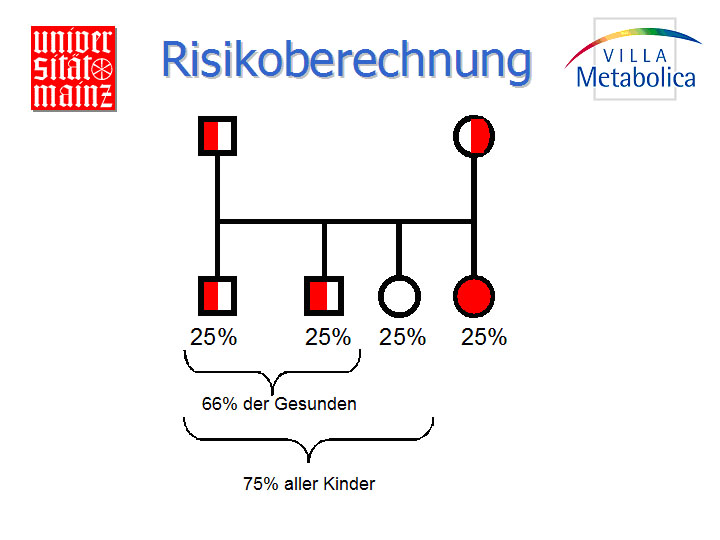
Das Wiederholungsrisiko bei dem autosomal-rezessiven Erbgang in einer betroffenen Familie ist aus den Mendelschen Regeln abzuleiten. In Partnerschaften, in denen beide Partner heterozygote Überträger (Konduktoren) sind, besteht bei jeder eintretenden Schwangerschaft ein 25%-Risiko, dass beide Partner ihre funktionslosen mutanten Gene an das Kind weitergeben, das daher manifest erkrankt. Die Wahrscheinlichkeit, dass nur einer der beiden Partner sein funktionsloses Gen überträgt, wodurch das Kind genau wie seine Eltern zu einem heterozygoten Konduktor wird, beträgt 50%. Mit einer Wahrscheinlichkeit von 25% übertragen schließlich beide Eltern ein intaktes Gen, so dass das Kind weder erkrankt noch zum Konduktor wird. Die Nachkommen von heterozygoten Eltern werden zu 25% betroffen sein, 75% sind gesund, davon sind jedoch 2/3 ebenfalls heterozygot. Der Heterozygoten-Status weiterer Verwandten lässt sich nur durch Gen-Analyse feststellen, wobei jedoch die Mutation des Index-Falles bekannt sein muss. Durch Enzym-Messung lässt sich ein Heterozygoten-Status nicht mit Sicherheit feststellen. Es besteht keine strenge Genotyp/Phänotyp-Beziehung, im Einzelfall können der Schweregrad und der Verlauf einer Erkrankung nicht vorhergesagt werden.
Drei lysosomale Speicherkrankheiten folgen dem X-chromosomalen Erbgang: Es sind dies die Mukopolysaccharidose Typ II (Morbus Hunter), der M. Fabry und der M. Danon.
Dem M. Hunter, einer X-chromosomal erblichen Erkrankung, liegt eine Defizienz der Iduronat-Sulfat-Sulfatase zugrunde. Schwere und leichte Krankheitsverläufe sind beschrieben worden, wobei die Extremformen als Typ A und Typ B bezeichnet werden: Der schwere Typ A ist in seinem klinischen Bild mit dem M. Hurler vergleichbar, wobei jedoch eine Hornhaut-Trübung fehlt. Der Typ B ist gekennzeichnet durch Gelenk-Kontrakturen, Schwerhörigkeit und Organvergrößerung bei normaler intellektueller Entwicklung. Während die häufigen Leistenhernien keine schwere Beeinträchtigung bedeuten, stellt die kardiale Manifestation (Aorten- und Mitralinsuffizienz durch verdickte Klappen) eine deutliche Gefährdung für den Patienten dar. Eine weitere Komplikation im späteren Lebensalter steht die zunehmende Sehverschlechterung dar, bei der Fundoskopie stellt sich das Bild einer Retinitis pigmentosa dar.
Der Morbus Fabry ist ebenfalls eine x-chromosomal vererbte lysosomale Speichererkrankung, die durch einen Mangel des Enzyms α-Galaktosidase verursacht wird und zu pathologischen Ablagerungen von Sphingolipiden in Endothelzellen, glatter Gefäßmuskulatur und dem Parenchym verschiedener Organe führt. Heterozygote Überträgerinnen sind oft ebenfalls manifest erkrankt.
Der Morbus Danon ist erst vor einem Jahrzehnt zum ersten Mal beschrieben worden: Diese lysosomale Speicherkrankheit beruht auf dem Defekt eines für die Funktion des Lysosom wesentlichen Membran-Proteins, des LAMP-2 (lysosomal associated membrane protein 2). Das klinische Bild ist gekennzeichnet durch eine bereits in früher Jugend einsetzende Kardiomyopathie mit Rhythmusstörungen und eine Myopathie. Wie bei M. Fabry ist auch bei M. Danon das weibliche Geschlecht betroffen.
Es stellt sich nun die Frage, warum Frauen, die das mutierte Gen für den M. Hunter tragen, lediglich Carrier ohne klinische Symptome darstellen, während Heterozygote mit M. Fabry und M. Danon manifest erkranken.
Die klinische Manifestation des M. Fabry beim weiblichen Geschlecht wurde von vielen Autoren mit einem Ungleichgewicht in der X-Inaktivierung des mutierten und normalen X-Chromosoms in Verbindung gebracht. Diese Hypothese konnte jedoch durch eine Studie von Maier et al. nicht bestätigt werden [1]. Dobyns und Mitarbeiter haben in einer umfangreichen Literatur-Übersicht 32 X-chromosomal erbliche Erkrankungen hinsichtlich der Penetranz und Expression der Symptome bei den weiblichen Individuen analysiert [2]. Sie konnten nachweisen, dass bei vielen dieser Krankheiten die Frauen in unterschiedlichem Maße symptomatisch sind, wobei die Penetranz der Symptome bei den verschiedenen Krankheiten große Unterschiede aufweist, sie reicht von 10% (Hämophilie A und B) bis 100% (Vitamin D-resistente Rachitis). Zur Erklärung dieses Phänomens ist es erforderlich, Überlegungen zum Gen-Produkt anzustellen: Kann das Gen-Produkt zwischen den Zellen ausgetauscht werden (wie bei M. Hunter), oder ist es Zellautonom, das heißt, der Gen-Defekt in einer Zelle kann nicht durch eine andere Zelle kompensiert werden, das Gen-Produkt ist Zellautonom. Ein Beispiel dafür ist die Vitamin-D resistente Rachitis. Die klinische Manifestation bei Überträgerinnen (Heterozygoten) des M. Danon lässt sich ebenfalls damit erklären, dass das Gen-Produkt, ein Membran-Protein, nicht zwischen Zellen ausgetauscht werden kann. Bei M. Fabry scheint noch ein anderer Faktor eine Rolle zu spielen: Aerts und Kollegen konnten nachweisen, dass bei dieser Sphingolipidose nicht nur Globotriaosylzeramid gespeichert wird, sondern auch das (wasserlösliche) Globotriaosylsphingosin [3]. Und diese Substanz hemmt die Aktivität der α-Galaktosidase. Als Schlussfolgerung ergibt sich aus diesen Überlegungen, dass jede X-chromosomale Krankheit hinsichtlich der Pathogenese und Penetranz beim weiblichen Geschlecht untersucht werden muss. Die Begriffe rezessiv und dominant sollten hier keine Anwendung mehr finden.

Abb. 1: Risikoberechnung.

Abb. 2: Lyosome-associated membrane protein 2.

Abb. 3: Penetranz des Phänotyps bei Frauen.
Literatur
-
Maier EM et al. Disease manifestations and X inactivation in heterozygous females with Fabry disease. Acta Paediatr Suppl 2006;95:30-38
-
Dobyns WB et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. American journal of medical genetics 2004;129A:136-143
-
Aerts JM et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proceedings of the National Academy of Sciences of the United States of America 2008;105:2812-2817
Referent
Prof. Dr. med. Michael Beck,
Universitäts-Kinderklinik
Langenbeckstrasse 1
55101 Mainz
Quelle: Satelliten-Symposium der Firma Actelion zum Thema „Lyosomale Speichererkrankungen: selten gesehen – oft übersehen“ am 12.09.2008 in München, anlässlich der 104. Jahrestagung der Deutschen Gesellschaft für Kinder- und Jugendmedizin (CGC Cramer-Gesundheits-Consulting) (tB).