


MEDIZIN
AWARDS
Forschergeist gefragt: 14. Novartis Oppenheim-Förderpreis für MS-Forschung ausgelobt
FernstudiumCheck Award: Deutschlands beliebteste Fernhochschule bleibt die SRH Fernhochschule
Vergabe der Wissenschaftspreise der Deutschen Hochdruckliga und der Deutschen Hypertoniestiftung
Den Patientenwillen auf der Intensivstation im Blick: Dr. Anna-Henrikje Seidlein…
Wissenschaft mit Auszeichnung: Herausragende Nachwuchsforscher auf der Jahrestagung der Deutschen…
VERANSTALTUNGEN
Wichtigster Kongress für Lungen- und Beatmungsmedizin ist erfolgreich gestartet
Virtuelle DGHO-Frühjahrstagungsreihe am 22.03. / 29.03. / 26.04.2023: Herausforderungen in…
Pneumologie-Kongress vom 29. März bis 1. April im Congress Center…
Die Hot Topics der Hirnforschung auf dem DGKN-Kongress für Klinische…
Deutscher Schmerz- und Palliativtag 2023 startet am 14.3.
DOC-CHECK LOGIN
Lysosomale Speicherkrankheiten:
Eine Einführung
Von Prof. Dr. med. S. vom Dahl, Köln
Wiesbaden (16. April 2007) – Die etwa fünfzig bisher bekannten lysosomalen Speicherkrankheiten sind genetisch bedingte Stoffwechsel-Störungen. Zugrunde liegt in der Regel ein Enzymdefekt beim Abbau eines Sphingolipids oder Glykogen, der zur lysosomalen Speicherung des entsprechenden Lipids bzw. von Glykogen führt. Die lysosomalen Speicher-Krankheiten gehören zwar zu den seltenen Erkrankungen („orphan diseases"), sind aber unter den seltenen Stoffwechselkrankheiten mit einer Frequenz von 1 ‑ 3 pro 100.000 Einwohner recht häufig. Sie werden in der Regel autosomal‑rezessiv vererbt. Abortive Formen dieser Erkrankungen werden oft zu spät oder gar nicht diagnostiziert. Zwischen den ersten Symptomen und der Diagnose vergehen meist 5‑10 Jahre, u. a. auch, weil immer noch nicht allgemein bekannt ist, dass einige der lysosomalen Speicherkrankheiten inzwischen gut behandelbar sind. Hierzu gehören die Sphingolipidosen (u.a. M. Gaucher und M. Fabry), die Mukopolysaccharidosen (Typ I ‑ M. Hurler, Typ II ‑ M. Hunter sowie Typ VI M. Maroteaux‑Lamy) sowie die Glykogenose Typ II (M. Pompe). Pathophysiologisches Prinzip der derzeit verfügbaren etablierten medikamentösen Behandlungsformen dieser Erkrankungen ist entweder die Verhinderung der weiteren Akkumulation der jeweiligen Substanz (Substratregulationstherapie) oder die Förderung des Abbaus der jeweiligen Speichersubstanz (Enzymersatztherapie).
Beim adulten M. Gaucher liegen neben Panzytopenie und Hepatosplenomegalie schwere invalidisierende Knochenveränderungen vor. Diese Symptome können mit der intravenösen Enzymersatztherapie mit Imiglucerase behandelt werden. Innerhalb von mehreren Jahren kommt es zu einer Verbesserung oder Normalisierung der Organgröße und der Panzytopenie sowie zur Verbesserung des Knochenbefalls. Bei Kindern mit Gedeihstörungen führt die Enzymersatztherapie in der Regel zu einem Wachstumsschub. Die Prognose ist bei rechtzeitiger Behandlung gut. Die neurologischen Manifestationen der Erkrankung lassen sich mit der Enzymersatztherapie allerdings nicht behandeln.
Der M. Fabry manifestiert sich oft mit massiven Extremitätenschmerzen, ist in nicht seltenen Fällen der Grund für ein chronisches Nierenversagen, eine KHK oder einen Schlaganfall in jungen Jahren und kann ebenfalls mittels Enzymersatztherapie behandelt werden. Neben einer symptomatischen Besserung kann die renale Progression dieser Erkrankung oft aufgehalten werden.
Für die Mukopolysaccharidose Typ I ist eine rekombinante intravenöse Enzymersatztherapie zugelassen. Lungenfunktion, Hepatomegalie und Gelenkbeweglichkeit der kleinen Patienten werden hierdurch verbessert, die intellektuellen Defizite lassen sich jedoch nicht beeinflussen.
Gute Erfolge hat eine Enzymersatztherapie auch beim M. Pompe, der Glykogenose Typ II. Früh eingesetzt, können schwere Muskelhypotonie („floppy infant") und respiratorisches Versagen vermieden werden.
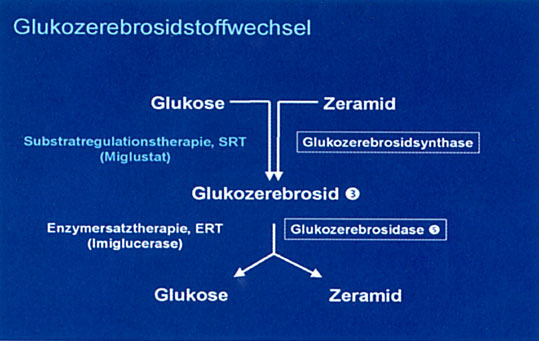
Die Substratregulationstherapie mit Miglustat, d.h. die Verhinderung der weiteren Akkumulation des bei M. Gaucher, aber z.B. auch bei M. NiemannPick Typ C gespeicherten Glykosphingolipids, stellt inzwischen eine Behandlungsalternative für Patienten mit M. Gaucher dar und befindet sich für die Indikation M. Niemann‑Pick Typ C in der klinischen Prüfung (Zulassungsantrag wurde bereits eingereicht). Da Miglustat ein so genanntes „kleines Molekül" ist, passiert es die Blut‑Hirn‑Schranke. Möglicherweise könnten damit auch neurologische Symptome, die sich mit der Enzymersatztherapie nicht behandeln lassen, in Zukunft therapiert werden. Bei Patienten mit M. Gaucher scheint die Substratregulationstherapie vor allem Vorteile hinsichtlich der Therapie von Knochensymptomen und Knochenschmerzen zu bieten.
Die Erfassung von Daten zur Wirksamkeit und/oder Sicherheit von „orphan drugs" in krankheitsspezifischen Registern ist in manchen Ländern obligat, z.B. auch im Rahmen von Zulassungsauflagen für neue Substanzen. Diese Register sind eine große Hilfe bei der Gewinnung neuer Erkenntnisse über seltene Erkrankungen und ihre Therapie auch im Langzeitverlauf.
Keyslides des Vortrages:

Abb. 1: Glukozerebrosidstoffwechsel.

Abb. 2: EU Orphan Drug Act 2000.

Abb. 3: Erste Erfahrungen mit Miglustat in Deutschland.

Abb. 4: Erfahrungen mit Miglustat nach 3 Monaten.
Quelle: Satelliten-Symposium der Firma Actelion Pharmaceuticals Deutschland zum Thema “Lyosomale Speicherkrankheiten im Fokus – viele Gesichter einer Gruppe seltener Erkrankungen“ anlässlich des 113. Kongresses der Deutschen Gesellschaft für Innere Medizin (DEGIM) am 16.04.2007 in Wiesbaden (CGC – Cramer-Gesundheits-Consulting).